

鼠库全书
-
-

-
代谢小鼠模型中的糖脂代谢紊乱模型如何构建?有哪些考量因素?
2025-02-13在代谢性疾病的研究领域内,相对于基因编辑小鼠用于研究单一基因对代谢功能影响的研究,饲料喂养构建的代谢小鼠模型更具备高临床模拟性,也更适合于研究代谢性疾病的发展过程。然而,此类小鼠模型的造模周期长,造模细节多,任何环节控制不好都无法获得预期的表型,因此,掌握有效造模方式,可以助力科研事半功倍。今天,亚星yaxing868官网就以糖脂代谢紊乱模型的构建为例,带你了解相关考量因素。
-
-
-
-
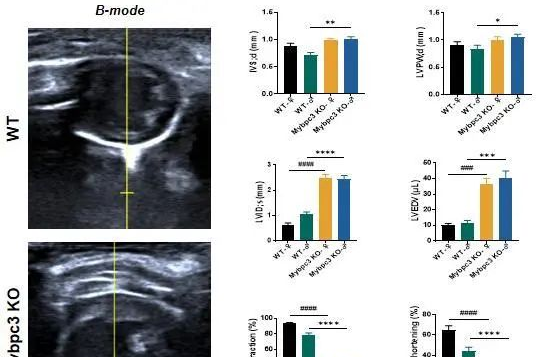
小身材,大能量——肥厚型心肌病小鼠模型助力药物开发
2025-02-12肥厚型心肌病(HCM)最常见的心肌病,患病率在1/200-1/500[1,2],其主要特征是心室壁增厚、心脏功能下降,引发进行性呼吸困难、心绞痛、心力衰竭、心房颤动和最可怕的心源性猝死。当前的治疗方法可以改善部分患者的症状,但它们并没有解决潜在的疾病病理生理学问题。这种未满足的临床需求迫切需要新的治疗手段可以从病理机制层面改善心肌细胞的舒张缺陷功能从而治疗HCM。HCM同样是最常见的遗传性心脏病,大约55%的HCM与基因突变相关,其中最常见的突变均发生在肌节蛋白和相关肌丝蛋白基因中。Mybpc3和Myh7是HCM中突变率最*高的两个蛋白,两者突变率之和几乎占比70%因基因突变导致的HCM[3,4]。着眼于肌节蛋白新型治疗方法为治愈HCM带来了新的希望。
-
-
-
-
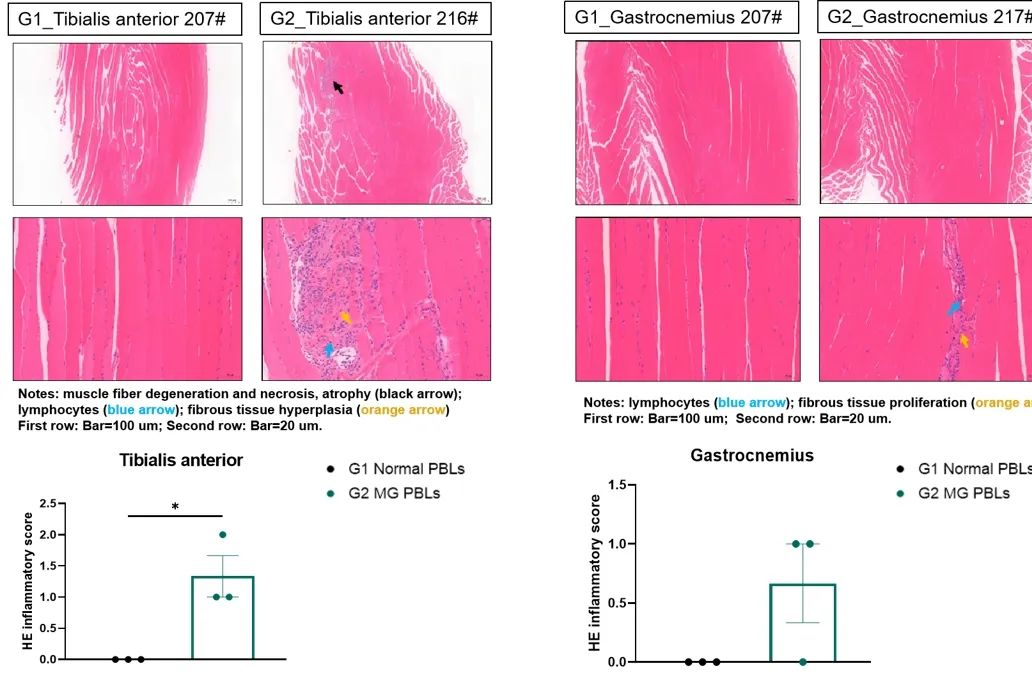
开启“有力”人生——免疫系统重建小鼠模型助力重症肌无力疾病新药研发
2025-02-11什么是重症肌无力疾病?它的致病机制如何?在当前的治疗现状下,亚星yaxing868官网免疫系统重建小鼠模型又将助力该疾病的新药研发?本文带你了解。
-
-
-

-
什么是渐冻症?亚星yaxing868官网渐冻症小鼠模型如何助力“融化”渐冻症?
2025-01-23“世界渐冻人日”的设立旨在提升公众对渐冻症的了解与关注。自1997年起,“渐冻人”协会国际联盟将每年的6月21日定为世界渐冻人日,这一天恰好是北半球一年中日照时间最长的一天,寓意着盛夏的阳光可以消融冰雪,为渐冻症患者带来希望。那么,什么是渐冻症?亚星yaxing868官网渐冻症小鼠模型如何在渐冻症的新药开发中发挥作用?本文带你了解。
-
-
-

-
难以模拟人类复杂疾病和基因调控?亚星yaxing868官网野化鼠来帮忙
2025-01-17实验室近交系小鼠有何局限性?为什么野化鼠更适合模拟人类复杂疾病和基因调控?亚星yaxing868官网为您解读。
-
-
-
-
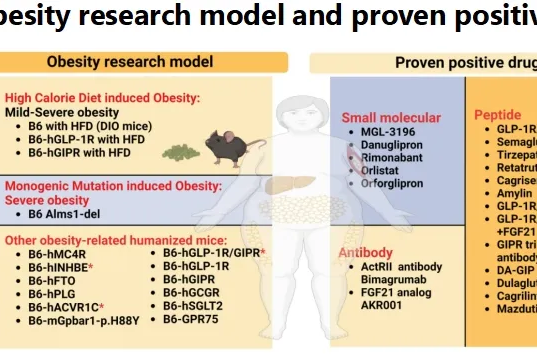
民为生物两款三靶点1类新药获得临床试验默示许可!解锁肥胖小鼠模型非临床评价策略
2025-01-15近日,中国国家药监局(NMPA)药品审评中心(CDE)官网公示,由民为生物申报的2款1类新药MWN109注射液,MWN105注射液获批临床试验(IND)许可,这两款新药均拟开发治疗II型糖尿病、超重或者肥胖。根据已公开资料,MWN109注射液,GIP/GLP-1/GCG三靶点脂肪酸链修饰多肽药物;MWN105注射液,GIP/GLP-1/FGF21三靶点Fc融合蛋白药物,目前两个项目都处于一期临床阶段。
-